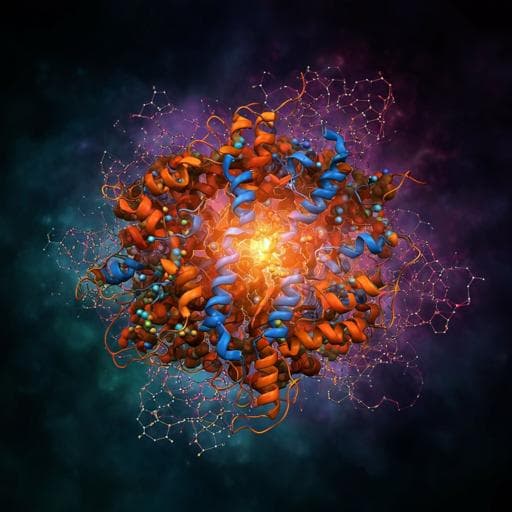
ChemistryNature Chemistry
Directed ultrafast conformational changes accompany electron transfer in a photolyase as resolved by serial crystallography
A. Cellini, M. K. Shankar, et al.
Delve into the fascinating world of charge-transfer reactions in proteins with groundbreaking research from Andrea Cellini, Madan Kumar Shankar, and team. Their study reveals the intricate structural dynamics involved in electron transfer processes within the *Drosophila melanogaster* photolyase, highlighting the role of conserved tryptophans and other key residues, and challenging longstanding theories in the field.
Related Publications
Explore these studies to deepen your understanding
Adjacent work that informs or extends this paper's methodology and findings.

Education
Thai Menschenbild: A Study of Chinese, Thai, and International Students in a Private Thai University as measured by the National Survey of Student Engagement (NSSE)
T. Waters and M. J. Day
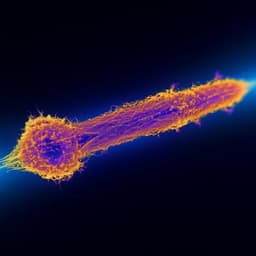
Physics
Transient lensing from a photoemitted electron gas imaged by ultrafast electron microscopy
O. Zandi, A. E. Sykes, et al.

Environmental Studies and Forestry
S-ZVI@biochar constructs a directed electron transfer channel between dechlorinating bacteria, *Shewanella oneidensis* MR-1 and trichloroethylene
H. Lyu, H. Zhong, et al.

Chemistry
Designed Rubredoxin miniature in a fully artificial electron chain triggered by visible light
M. Chino, L. F. D. Costanzo, et al.


