Medicine and HealthNature Communications
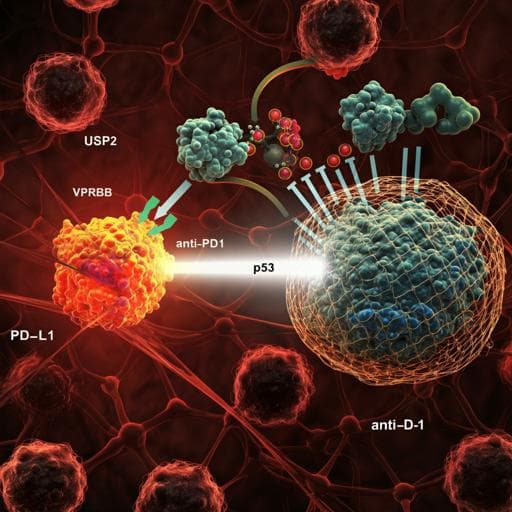
Targeting USP2 regulation of VPRBP-mediated degradation of p53 and PD-L1 for cancer therapy
J. Yi, O. Tavana, et al.
Exciting discoveries in cancer therapy show that targeting the USP2-VPRBP axis can effectively reactivate p53 and reduce toxicity compared to traditional Mdm2 inhibitors. This innovative approach, explored by authors Jingjie Yi, Omid Tavana, Huan Li, Donglai Wang, Richard J. Baer, and Wei Gu, demonstrates complete tumor regression in wild-type p53 tumors with the combination of a USP2 inhibitor and anti-PD1 antibody.
Related Publications
Explore these studies to deepen your understanding
Adjacent work that informs or extends this paper's methodology and findings.

Medicine and Health
Radiogenomics for predicting p53 status, PD-L1 expression, and prognosis with machine learning in pancreatic cancer
Y. Iwatate, I. Hoshino, et al.
Engineering and Technology
Nanoparticles and convergence of artificial intelligence for targeted drug delivery for cancer therapy: Current progress and challenges
R. P. Singh, A. Natarajan, et al.

Medicine and Health
Efficacy and safety of serplulimab plus nab-paclitaxel in previously treated patients with PD-L1-positive advanced cervical cancer: a phase II, single-arm study
J. An, X. Li, et al.
Medicine and Health
Advances in Photodynamic Therapy for the Treatment of Actinic Keratosis and Nonmelanoma Skin Cancer: A Narrative Review
A. S. Farberg, W. Justin, et al.


