Medicine and HealthNature Communications
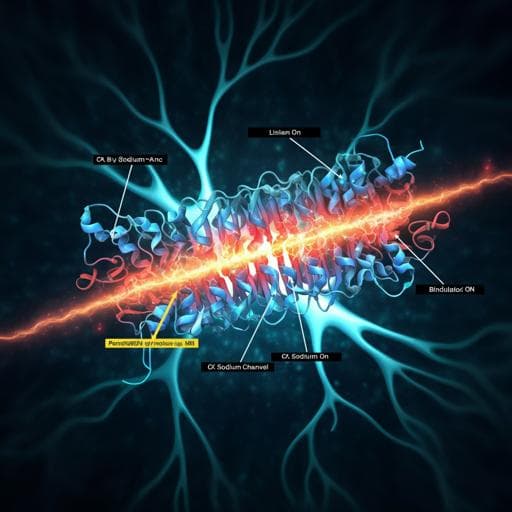
Structural basis for modulation of human Nav1.3 by clinical drug and selective antagonist
X. Li, F. Xu, et al.
Dive into the fascinating world of voltage-gated sodium channels! This study reveals how NaV1.3 interacts with specific modulators like bulleyaconitine A and ICA121431, shedding light on their unique binding mechanisms. Conducted by esteemed researchers including Xiaojing Li and Feng Xu, these findings are paving the way for targeted therapeutic advancements.
Related Publications
Explore these studies to deepen your understanding
Adjacent work that informs or extends this paper's methodology and findings.
Biology
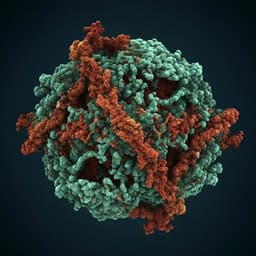
Structural basis for the activation of plant bunyavirus replication machinery and its dual-targeted inhibition by ribavirin
J. Li, L. Cao, et al.

Medicine and Health
A comparative study of COVID-19 transcriptional signatures between clinical samples and preclinical cell models in the search for disease master regulators and drug repositioning candidates
H. Chapola, M. A. D. Bastiani, et al.
Medicine and Health
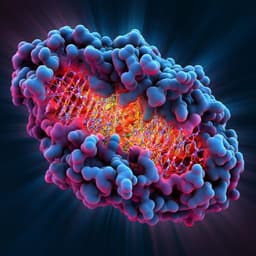
Discovery of a selective and biologically active low-molecular weight antagonist of human interleukin-1β
U. Hommel, K. Hurth, et al.
Medicine and Health
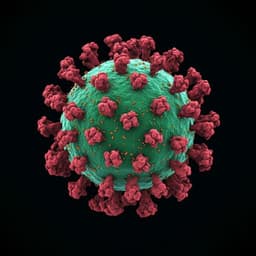
Structural insights into the modulation of coronavirus spike tilting and infectivity by hinge glycans
D. Chmielewski, E. A. Wilson, et al.


