Medicine and HealthNature Communications
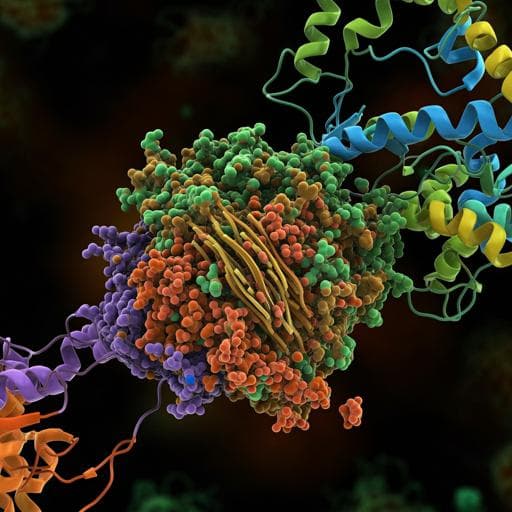
Molecular insights into receptor binding energetics and neutralization of SARS-CoV-2 variants
M. Koehler, A. Ray, et al.
This cutting-edge study reveals how SARS-CoV-2 mutations, particularly N501Y and E484Q, modify receptor binding and antibody neutralization. By employing atomic force microscopy and molecular dynamics, the authors delve into the stability of the RBD-ACE2 complex, offering crucial insights into the variant's implications on immunity. This research was conducted by Melanie Koehler, Ankita Ray, Rodrigo A. Moreira, Blinera Juniku, Adolfo B. Poma, and David Alsteens.
Related Publications
Explore these studies to deepen your understanding
Adjacent work that informs or extends this paper's methodology and findings.

Medicine and Health
Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants
P. Han, C. Su, et al.

Medicine and Health
Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor
J. Yang, S. J. L. Petitjean, et al.
Medicine and Health
Human antibodies to SARS-CoV-2 with a recurring YYDRXG motif retain binding and neutralization to variants of concern including Omicron
H. Liu, C. I. Kaku, et al.
Medicine and Health
Broad host range of SARS-CoV-2 and the molecular basis for SARS-CoV-2 binding to cat ACE2
L. Wu, Q. Chen, et al.


