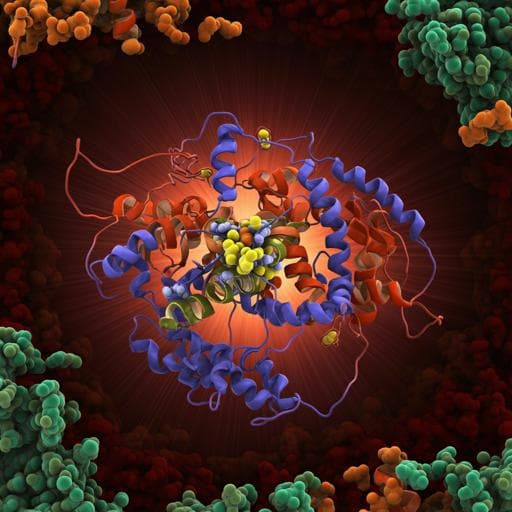
Key role of quinone in the mechanism of respiratory complex I
J. Gutiérrez-fernández, K. Kaszuba, et al.
Explore these studies to deepen your understanding
Adjacent work that informs or extends this paper's methodology and findings.

Social Networks Play a Complex Role in HIV Prevention Knowledge, Attitudes, Practices, and the Uptake of PrEP Through Transgender Women Communities Centered Around Three "Casas Trans" in Lima, Peru: A Qualitative Study
T. Temelkovska, K. Moriarty, et al.

Role of job mobility frequency in job satisfaction changes: the mediation mechanism of job-related social capital and person-job match
H. Yang and P. Hu

The influence of the parental child-rearing gender-role attitude on children's social adjustment in single- and two-parent families: the mediating role of intergenerational identity
I. Chen, Y. Wang, et al.
Role of meteorological factors in the transmission of SARS-CoV-2 in the United States
Y. Ma, S. Pei, et al.


